В
последние 20 лет произошли
существенные сдвиги в изучении и
лечении так называемых липидогенных, форм
ускоренного атеросклероза. Наиболее значительны событиями
явились массовая первичная
профилактика эпидемии
атеросклероза с помощью коррекции
важнейших
риск-факторов, а также создание
целого арсенала гиполипидемических препаратов для
вторичной профилактики инфаркта
миокарда. В настоящее время трудно
себе представить современную
кардиологическую клинику без статинов,
фибратов, никотиновой кислоты
и липофильныхантиоксидантов. Однако
количество операций коронарного шунтирования и ангиопластики быстро
увеличивается во всех странах.
Следовательно, кардиологи пока не
имеют эффективных лекарств против ишемической болезни сердца (ИБС) и коронарного
атеросклероза. Опыт многих стран
показывает, что около трети
пациентов с ИБС оказались резистентны к ловастатину и комбинациям
гиполипидемических препаратов. В
группе высочайшего риска оказались
дети, страдающие так называемой
семейной гипер-холестеринемией и гиперлипопротеинемией III типа. Цель
лекции - рассказать о клеточной
терапии этих наследственных
моногенных заболеваний с помощью
донорских аллогенныхгепатоцитов и стволовых
гематогенных клеток. Хотя эти
исследования только начинаются в
Европе и США, они знаменуют новую
эпоху и новое мышление в лечении
атеросклероза, так как впервые
предпринимается попытка
корригировать атерогенную
дислипопротеидемию путем
"микротрансплантации" и коррекции
клеточного состава органов больных
людей.
Семейная гиперхолестеринемия(СГ) является одним
из хорошо изученных заболеваний липопротеидного обмена,
обусловленным доминантной
мутацией гена, ответственного за
синтез мембранного рецептора.
Специфическим лигандом этого
рецептора являются липопротеиды низкой
плотности (ЛНП), которые в кровотоке являются
главными транспортерами пищевого, атеро-генного холестерина. 75% ЛНП-рецепторов в организме
человека сосредоточено на
поверхности гепатоцитов. Важнейшая
физиологическая функция
ЛНП-рецепторов - быстрое
эффективное удаление ЛНП в печень,
где избыток холестерина окисляется
в желчные кислоты и выводится из
организма через кишечник. В других
соматических клетках и тканях ЛНП-рецепторы
слабо
экспонированы из-за высокого
содержания холестерина в клетках.
Ген ЛНП-рецептора локализован на
19 хромосоме человека. Этот мультидоменный мембранный
белок имеет множество функций.
Описана топография делеций и мутаций гена,
ведущих к функциональным
повреждениям разных участков
рецептора. (Рис 1). Большинство
делеций заканчивается
прекращением синтеза мРНК и белка, тогда
как точечные мутации ведут к
синтезу функционально
неполноценного рецептора. У пациентов-гетерозигот 50% — снижение
активных ЛНП-рецепторов приводит к
двукратному повышению уровня ЛНП в
плазме крови, раннему развитию ИБС,
коронарного атеросклероза с
инфарктом миокарда в первой
половине жизни. Частота
встречаемости таких пациентов 1 на
500 жителей. Гомозиготы встречаются
весьма редко (1 на миллион жителей).
У гомозигот СГ уровень
холестерина достигает 1000 мг/дл, в 15-25 лет
развивается тяжелый коронарный
атеросклероз. Без трансплантации
печени гомозиготная форма
заболевания заканчивается
летально. Более того,
трансплантированная печень
функционирует у таких больных не
более 1-2 лет (причины неудачных
пересадок пока детально не изучены)
(1). Тяжесть и скорость развития
коронарного атеросклероза весьма
варьируют среди гетерозигот СГ. Наиболее
существенную роль играет
полиморфизм гена, контролирующего
синтез аполипопротеинаЕ. Этот белок
контролирует альтернативный путь
выведения пищевых липидов и холестерина в
печень.
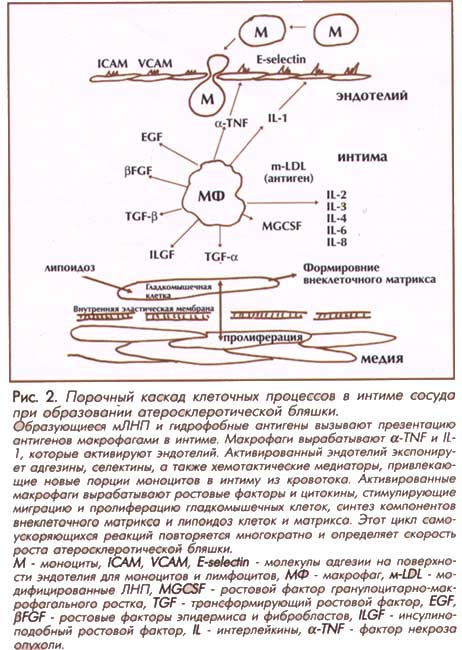
У больных СГ
отчетливо прослежена взаимосвязь
между высоким уровнем ЛНП в плазме
крови и формированием атероскле-ротических бляшек в
коронарных артериях (Рис 2). Из-за
сниженного клиренса в печени все
пищевые липопротеиды очень низкой
плотности (ЛОНП) успевают
превратиться в ЛНП, которые
остаются циркулировать в
кровотоке. У здоровых людей лишь
половина ЛОНП превращается в ЛНП. У
больных концентрация и время
пребывания ЛНП в плазме крови
нарастают. В результате возрастает
число частиц ЛНП, которые
фильтруются в интиму артерий. В интиме часть ЛНП
подвергается окислению и
преобразованиям в
модифицированные частицы (м-ЛНП). Некоторые мЛНП вызывают
образование новых антигенов и
стимулируют их представление
макрофагами. Немаловажно и то, что
мЛНП приносят в интиму чужеродные
антигены (липополисахаридызапускает
бактерий, ксенобиотики). Активация макрофагов
воспалительную и иммунную
активацию эндотелия и приводит к
повышению экспрессии молекул
адгезии (ICAM, VCAM, E-selectins) на
поверхности эндотелиальныхадгезинов и клеток,
контактирующих с кровью. Активация
медиаторов хемотаксиса привлекает
новые популяции моноцитов и
лимфоцитов. Активированный
эндотелий и макрофаги интиму секретируют
в
ростовые факторы и пролиферацию
гладкомышечных клеток (ГМК), синтез экстра-целлюлярногоцитокины, ускоряющие
миграцию и матрикса (фиброз) и
отложение новых порций ЛНП и мЛНП
(липоидоз). Этот "порочный
круг" повторяется в интиме много раз с
усилением, что вызывает рост
атеросклеротической бляшки.
Нерегулируемый рост бляшки в
интиме напоминает феномены "клеточного
хаоса", которые
наблюдаются при циррозе печени,
доброкачественной гиперплазии
простаты и других клеточных дисплазиях (4). В настоящее
время единственным способом
блокирования роста бляшек является
их химическое или физическое
удаление, либо механическое раздавливание(ангиопластика), а единственным
эффективным лечением гомозигот - пересадка
печени, которая восстанавливает
нормальный круговорот пищевого
холестерина в организме. Ловастатин на 20-60% снижает
уровень ЛНП у ге-терозигот, селективно
повышая уровень ЛНП-рецепторов в печени.
Однако не менее половины пациентов
плохо переносят ловастатин (это
лекарство назначается до конца
жизни, т.к. прекращение его приема
приводит к 2-3-кратному повышению
холестерина плазмы крови).
Результаты многочисленных
проектов "генной терапии" СГ пока также не
обнадеживают. Технологию пересадок
ген-трансфецированныхгепатоцитов удалось
частично отработать на кроликах Ватанаби (эти кролики
оказались весьма точной гено- и фенокопией СГ человека), но
генная терапия СГ не станет в
ближайшее время клинической
реальностью. Что удерживает
родителей и врачей от этой дорогой
и весьма рискованной операции?
Во-первых, у тяжело больного
ребенка нужно под [beep]зом иссечь
треть печени, чтобы выделить около
10 млрд гепатоцитов. На этапе
изолирования и получения первичной
культуры гепатоцитов теряются
около 3 млрд клеток. Оставшиеся 7
млрд гепатоцитов шуиго модифицируют,
используя в качестве вектора мутантныйаденовирус. В качестве
"начинки" используют два гена -
ЛНП-рецептор и ген бета-галактозидазы
(активность
которого в клетках можно
контролировать с помощью хромогенного субстрата).
Максимальная эффективность трансфеции in vitro равна 40-30%.
Следовательно, пациенту вновь
возвращаются в печень только 3.5
млрд клеток с восстановленным ЛНП-рецептором от исходных 10
млрд клеток. Однако самым слабым
звеном лечения является последний
этап возвращения гепатоцитов в
печень шприцевой инъекцией.
Эффективность приживляемоститрансфецированных клеток в печени
больных остается на уровне 1-2%, т. е. вся операция
завершается возвращением в печень
пациента 35 млн "вылеченных"
клеток. Остальные гепатоциты быстро
элиминируются из кровотока и
погибают, если даже после операции
использовать иммуносу-прессанты. Таким образом,
эффективность "генной
терапии" пока выражается
следующим отношением: одна
"вылеченная" клетка на 1000
"невылеченных". По этой
причине в США, Канаде, Франции и
Англии проведены только несколько
трансплантаций гепатоцитов после
их "генной коррекции" in vitro. У
всех пациентов сразу после
операции отмечался гиполипидемический эффект, который
сохранялся несколько недель.
Однако он был непродолжительным,
поскольку трансфецированные клетки
постепенно погибали, либо
переставали функционировать. Не
нужно забывать, что на их
поверхности остаются фрагменты аденовируса, которые
являются мощными антигенами.
Существуют также проблемы с
устойчивой длительной экспрессией
трансфецированных генов.
Предпринимаются
многочисленные попытки создать
генетические конструкции с
переносимым геном ЛНП-рецептора, которые смогли
бы при введении в кровоток эффективно трансфеци-ровать гепатоциты
человека. Первые положительные
результаты уже получены.
Одновременно
с генной терапией СГ стал
обсуждаться проект пересадки аллогенныхфетальных гепатоцитов. В
чем преимущество этого подхода?
Во-первых, не нужны гепатэктомия и тяжелая
операция под [beep]зом для больных
детей. Во-вторых, фе-тальные гепатоциты
более доступны, сеансы
трансплантации можно варьировать
по дозам и органам, а также
повторять неоднократно. В-третьих,
комплексы гистосовместимости еще слабо
выражены на фетальных гепатоцитах, реакции
отторжения легче контролировать.
В-четвертых, фетальные гепатоциты
сохраняют высокую потенцию к
пролиферации в присутствии
соответствующих ростовых факторов.
В-пятых, для лечения моногенных
метаболических дефектов
достаточна пересадка гепатоцитов
вместо пересадки печени.
Работы по
трансплантации нормальных
аллогенных гепатоцитов показали,
что для достижения необходимого гиполипидемиче-ского эффекта
требуется однократная пересадка 400
млн гепатоцитов. Это
свидетельствовало о низкой
приживляемости донорских
гепатоцитов в организме реципиента
и заставило отказаться от
однократного введения донорских
гепатоцитов в печень, сальник или
селезенку.
Более
устойчивый гиполипидемический
эффект давали аллоген-ные гепатоциты,
выращенные на микросферах, которые
пересаживали в брюшную полость. В
этих случаях пересадка нескольких
десятков миллионов гепатоцитов
давала хороший гиполипидемический
эффект в течение 10 дней. Применяя иммуносупрессанты, гиполипидемическое действие
пересаженных гепатоцитов
удавалось продлить на два и более
месяца. Неожиданным здесь было то,
что трансплантация аллогенных гепатоцитов в
брюшную полость стимулировала
захват ЛНП"больной" печенью.
Следовательно, аллогенныегепатоциты активируют
альтернативные пути катаболизма
ЛНП в печени при СГ. Еще более
перспективной выглядит пересадка
аллогенных фетальных гепатоцитов,
поскольку они способны формировать
в печени реципиента новые балки и
дольки. Уровень приживления фетальных
гепатоцитов можно оценивать по альфа-фетопротеину в плазме крови.
В настоящее время исследования с гепатоцитамифетальной печени активно
ведутся в нескольких медицинских
центрах США и Франции. Ведутся они и
в Кардиологическом Научном Центре
в Москве.
В
нескольких лабораториях было
показано, что введение аполи-попротеинаЕ резко
уменьшало гиперхолестеринемию и симптомы
коронарного атеросклероза у
животных с пищевой гиперхо-лестеринемией и кроликов Ватанаби. Более того, у
кроликов и мышей с трансгенноэкспрессированным высоким апоЕ
полностью блокировалось развитие алиментарнойгиперхолестерине-мии и
атеросклероза. Эти обнадеживающие
находки на животных заставили
предположить, что с помощью апо Е
удается усилить отток "атерогенных"липопротеидов
из крови в печень. Дальнейшие
исследования подтвердили эту
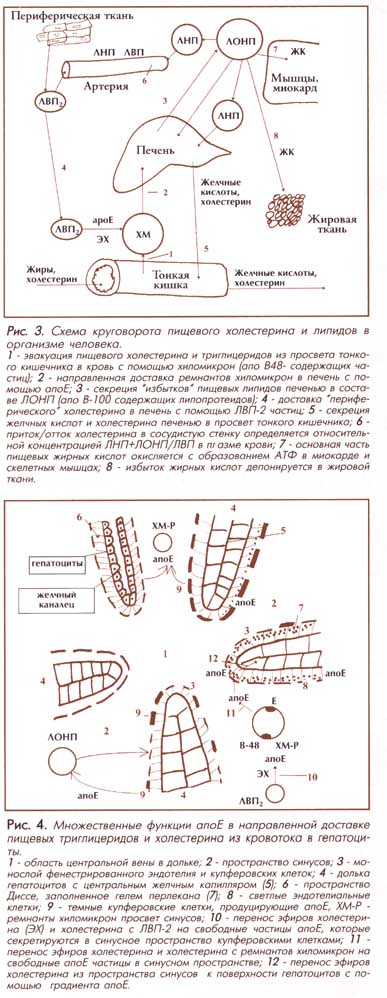
гипотезу. Как показано на Рис. 3,
апоЕ осуществляет независимую
доставку ремнантов хиломикрон (ХМ) и богатых апоЕ
липопротеидов высокой плотности (ЛВП) в печень.
Передача холестерина и пищевых
липидов с ремнантов ХМ в гепатоциты
осуществляется в два этапа. На
первом этапе в синусных
пространствах идет перенос
холестерина и его эфиров с
ремнантов и ЛОНП на апоЕ, который
синтезируется купферовскими
клетками. Убедительно доказано, что
в ответ на захват ремнантов ХМ
макрофаги синусной выстилки
синтезируют и секретируют в синусное
пространство апоЕ диско-идные частицы,
лишенные холестерина. Далее пул
новообразованных апоЕ,
продуцируемых купферовскими
клетками, обеспечивает "эстафетную" перекачку
холестерина и его эфиров из синусов
в пространство Диссе, как показано на
Рис. 4. Высокий градиент апоЕ,
продуцируемый гепатоцитами, удерживается в
гидрофильных гелях перлекана
(преобладающего гепарансульфа-та) в
пространствах Диссе. Особенностью
пространства Диссе является
отсутствие типичной базальной мембраны как на
синусных клетках, так и на
поверхности гепатоцитов,
обращенных в пространство Диссе.
Высокая концентрация апоЕ
сохраняется в пространстве Диссе
за счет высокоаффинных взаимодействий
апоЕ и перлекана. Этот белок имеет
специальный центр для селективного
связывания апоЕ белка. Деградированные мелкие частицы ремнантных комплексов
доставляются и удерживаются в
пространствах Диссе за счет высокоаффиных взаимодействий
ли-попротеидлипазы и гепарансульфата. Нарастающий
градиент апоЕ от
капилляров-синусов к пространству
Диссе обеспечивает направленный
транспорт холестерина в
гепатоциты. На этом основаны гиполипидемические эффекты моноцит-макрофаг-коло-нийстимулирующих факторов и
пересадки аллогенных стволовых
клеток. Эти процедуры усиливают
синтез перлекана в интер-стиции печени и
стволовых кроветворных
пространствах, куда "дренируются" лишние атерогенныелипопротеиды. Гепатоциты и
синусные клетки продуцируют апоЕ,
чтобы селективно "скачать" холестерин и
эфиры с крупных ремнантов ХМ и ЛОНП
на гепарансульфатную "оболочку"
гепатоцитов. Эта оболочка является
скоплением экстраклеточных
рецепторов для апоЕ содержащих липопротеидных частиц. В этом
случае ремнантам не нужно
пересекать синусные клетки, чтобы
оказаться в пространстве Диссе.
Окончательно челночные функции
апоЕ белка в переносе пищевых
липидов и холестерина в печень были
доказаны на трансгенных мышах,
которые синтезировали в 4-3 раз
больше апоЕ трансфецированными гепатоцитами.
Высокое содержание апоЕ в печени
трансгенных мышей позволило
зарегистрировать динамику
циклических перемещений апоЕ
частиц после приема жирной пищи и
при возвращении в "голодное" состояние.
Оказалось, что в голодном состоянии
основная часть "свободного"
апоЕ концентрируется в цитоплазме
вдоль цитоплазматической мембраны гепатоцитов, контактирующей
с областью синусов. При пищевых
нагрузках весь пул апоЕ переходит в
пространство Диссе, где
накапливается в перлекановом геле. Спустя 12-18
часов после пищевой нагрузки
основной пул апоЕ оказывается
вновь в цитоплазме. Таким образом,
перекачка липидов и холестерина
из пространства Диссе в гепатоциты осуществляется
циклической миграцией апоЕ частиц.
(3). Циклическая миграция апоЕ вдоль
плазматической мембраны
гепатоцитов выполняет роль
селективного "липидного канала". При высокой
концентрации апоЕ в перлекане и гепатоцитах этот
альтернативный путь перекачки
холестерина (в обход ЛНП рецепторов)
резко усиливается.
Пересадка аллогенных стволовых гемопоэтических клеток
вызывает появление новых популяций
купферовских клеток, активно
продуцирующих апоЕ в синусных
пространствах печени. Одновременно
размножающиеся аллогенные клоны
купферовских клеток активно
синтезируют перлекан, который
становится "полупроницаемым
ситом", на котором
задерживаются апоЕ содержащие липопротеидные частицы. Пересадка
стволовых гемопоэтических предшественников
оказалась также эффективным
методом терапии гиперлипопротеинемии III типа.
Отличительной особенностью этого
заболевания является нарушение
катаболизма хиломикрон и ЛОНП
вследствие
мутации в гене, контролирующем
синтез аполипопро-теинаЕ (синтезируется
неактивный белок апоЕ2 (или другие
молекулярные неактивные изоформы), которые не
способны образовывать прочные
комплексы с перлеканом. Большинство
пациентов, страдающих клинической
формой ускоренного коронарного
атеросклероза в сочетании с гиперлипопротеинемией III типа,
являются гомозиготными по мутантной аллели гена
Е2/Е2. Частота встречаемости Е2/Е2 или
других комбинаций функционально
дефектных апоЕ аллелей в
европейской популяции равна 1:100.
Однако реальная
распространенность заболевания
лишь 1:3000. Оказывается, для
проявления заболевания необходимы
такие факторы, как ожирение, диабет,
гипотиреоз и т.д. Эти
факторы изменяют скорость
всасывания липидов в кишечнике и
осложняют клиренсремнантовХМ в печени. О роли
гормонов свидетельствует тот факт,
что гиперлипопротеинемия III типа никогда
не развивается у мальчиков до
полового созревания и не бывает у
женщин до менопаузы.
Важным
диагностическим "маркером" СГ и гиперлипидемии III типа
являются множественные ксантомы,липидные полоски на
роговице и ксантелазмы. Наличие
дополнительных генов, влияющих на
распространенность и проявление
заболевания осложняло изучение
молекулярных механизмов болезни на
людях. Поэтому на мышах была
разработана упрощенная
генетическая модель заболевания,
когда обе аллели гена апоЕ были
выключены у мышей техникой knockout. У этих апоЕ-дефицитных мышей,
находившихся на обычной безлипидной диете, уровень
холестерина и триглицеридов поднимался до
430 мг/дл и 230 мг/дл,
соответственно. У взрослых мышей в интиме дуги и корней
восходящей аорты развивались
характерные липофиброзные разрастания
смешанных популяций клеток. (2, 3).
Опыты с трансплантацией печени
показали, что около 90% апоЕ в плазме
крови синтезируется гепатоцитами и только 1-2% -
макрофагами. Поскольку все
хозяйские клетки пациентов с
гиперлипопротеинемией III типа
продуцируют дефектный апоЕ, была
предпринята попытка восстановить
синтез нормального апоЕ с помощью
трансплантации аллогенных
стволовых гемопоэтических клеток
(6).
На первом
этапе эта идея экспериментально
отрабатывалась на апоЕ-дефектных трансгенных
мышах со спонтанным атеросклерозом
аорты. Было показано, что введение апоЕ4 в кровоток вызывало
мощный гиполипидемический эффект. Уровень
бета-ЛОНП и ЛОНП падал
почти до уровня контроля. На
следующем этапе апоЕ-дефицитным мышам
проводили рентгеновское облучение
дозой, вызвавшей гибель всего пула
собственных гемопоэтических
клеток. Далее части мышей проводили
пересадку костного мозга сингенных мышей с
нормальной экспрессией гена апоЕ.
Остальные мыши получали костный
мозг от линий с выключенным геном
апоЕ. Уже через 2 недели уровень
апоЕ у мышей-реципиентов,
получивших донорский костный мозг,
достиг уровня 12-13% от контроля. В
группе мышей, получивших аллогенный костный мозг с
выключенным геном апоЕ, в крови не
определялся апоЕ. На 3 и 4 неделе
после трансплантации уровень
холестерина снижался на 30 и 70%
соответственно. С помощью поточной цитофлуориметрии было
установлено появление популяции
моноцитов в кровотоке, которые в
культуре синтезировали апоЕ. Через
3 и 4 месяца после трансплантации
животные имели на порядок меньшие
области липидных отложений и
фиброзных возвышений интимы (2, 3). Таким
образом, донорские стволовые
клетки, пересаженные в
организм Животного с
генетическим дефектом апоЕ,
способны практически полностью
компенсировать биохимический
дефект за счет репопуляции донорских
клонов моноцитов-макрофагов в
организме реципиента.
Примечательно, что 10-12% апоЕ от
уровня апоЕ у контрольных Животных
оказывается достаточным для
коррекции ускоренного
атеросклероза. По-видимому, имеет
значение появление популяции апоЕ
синтезирующих моноцитов не только
в печени, но и зонах атеросклероти-ческих поражений аорты.
Результаты,
полученные в эксперименте,
оказались столь впечатляющими, что
в США от РОА и вашингтонской
администрации Национального
Института Здоровья выдано
разрешение на проведение
выборочных клинических испытаний
нового лечения. Сейчас в нескольких
клиниках США проходит независимая
апробация пересадки костного мозга
для коррекции наследственных атерогенныхдислипопротеидемий, угрожающих
преждевременной гибелью пациентов.
Не вызывает сомнений, что мы
вступаем в эпоху, когда многие
моногенные метаболические
расстройства, включая атероген-ныедислипопротедемии, начинают
лечиться не лекарствами и не
трансплантацией органов, а
трансплантацией стволовых и
соматических аллогенных клеток. В блиЖлйилие 10 лет область
таких вмешательств в
кардиологии будет беспрецедентно
расширяться.
Задачи фундаментальной
медицины заключаются в том, чтобы
устанавливая допустимые границы
таких вмешательств,
ответить на главный вопрос: кому, когда и
как необходима трансплантация
стволовых и дифференцированных
клеток для коррекции атерогенных
дислипопротеидемий.
 У больных СГ
отчетливо прослежена взаимосвязь
между высоким уровнем ЛНП в плазме
крови и формированием атероскле-ротических бляшек в
коронарных артериях (Рис 2). Из-за
сниженного клиренса в печени все
пищевые липопротеиды очень низкой
плотности (ЛОНП) успевают
превратиться в ЛНП, которые
остаются циркулировать в
кровотоке. У здоровых людей лишь
половина ЛОНП превращается в ЛНП. У
больных концентрация и время
пребывания ЛНП в плазме крови
нарастают. В результате возрастает
число частиц ЛНП, которые
фильтруются в интиму артерий. В интиме часть ЛНП
подвергается окислению и
преобразованиям в
модифицированные частицы (м-ЛНП). Некоторые мЛНП вызывают
образование новых антигенов и
стимулируют их представление
макрофагами. Немаловажно и то, что
мЛНП приносят в интиму чужеродные
антигены (липополисахаридызапускает
бактерий, ксенобиотики). Активация макрофагов
воспалительную и иммунную
активацию эндотелия и приводит к
повышению экспрессии молекул
адгезии (ICAM, VCAM, E-selectins) на
поверхности эндотелиальныхадгезинов и клеток,
контактирующих с кровью. Активация
медиаторов хемотаксиса привлекает
новые популяции моноцитов и
лимфоцитов. Активированный
эндотелий и макрофаги интиму секретируют
в
ростовые факторы и пролиферацию
гладкомышечных клеток (ГМК), синтез экстра-целлюлярного цитокины, ускоряющие
миграцию и матрикса (фиброз) и
отложение новых порций ЛНП и мЛНП
(липоидоз). Этот "порочный
круг" повторяется в интиме много раз с
усилением, что вызывает рост
атеросклеротической бляшки.
Нерегулируемый рост бляшки в
интиме напоминает феномены "клеточного
хаоса", которые
наблюдаются при циррозе печени,
доброкачественной гиперплазии
простаты и других клеточных дисплазиях (4). В настоящее
время единственным способом
блокирования роста бляшек является
их химическое или физическое
удаление, либо механическое раздавливание (ангиопластика), а единственным
эффективным лечением гомозигот - пересадка
печени, которая восстанавливает
нормальный круговорот пищевого
холестерина в организме. Ловастатин на 20-60% снижает
уровень ЛНП у ге-терозигот, селективно
повышая уровень ЛНП-рецепторов в печени.
Однако не менее половины пациентов
плохо переносят ловастатин (это
лекарство назначается до конца
жизни, т.к. прекращение его приема
приводит к 2-3-кратному повышению
холестерина плазмы крови).
Результаты многочисленных
проектов "генной терапии" СГ пока также не
обнадеживают. Технологию пересадок
ген-трансфецированных гепатоцитов удалось
частично отработать на кроликах Ватанаби (эти кролики
оказались весьма точной гено- и фенокопией СГ человека), но
генная терапия СГ не станет в
ближайшее время клинической
реальностью. Что удерживает
родителей и врачей от этой дорогой
и весьма рискованной операции?
Во-первых, у тяжело больного
ребенка нужно под [beep]зом иссечь
треть печени, чтобы выделить около
10 млрд гепатоцитов. На этапе
изолирования и получения первичной
культуры гепатоцитов теряются
около 3 млрд клеток. Оставшиеся 7
млрд гепатоцитов ш уиго модифицируют,
используя в качестве вектора мутантный аденовирус. В качестве
"начинки" используют два гена -
ЛНП-рецептор и ген бета-галактозидазы
(активность
которого в клетках можно
контролировать с помощью хромогенного субстрата).
Максимальная эффективность трансфеции in vitro равна 40-30%.
Следовательно, пациенту вновь
возвращаются в печень только 3.5
млрд клеток с восстановленным ЛНП-рецептором от исходных 10
млрд клеток. Однако самым слабым
звеном лечения является последний
этап возвращения гепатоцитов в
печень шприцевой инъекцией.
Эффективность приживляемости трансфецированных клеток в печени
больных остается на уровне 1-2%, т. е. вся операция
завершается возвращением в печень
пациента 35 млн "вылеченных"
клеток. Остальные гепатоциты быстро
элиминируются из кровотока и
погибают, если даже после операции
использовать иммуносу-прессанты. Таким образом,
эффективность "генной
терапии" пока выражается
следующим отношением: одна
"вылеченная" клетка на 1000
"невылеченных". По этой
причине в США, Канаде, Франции и
Англии проведены только несколько
трансплантаций гепатоцитов после
их "генной коррекции" in vitro. У
всех пациентов сразу после
операции отмечался гиполипидемический эффект, который
сохранялся несколько недель.
Однако он был непродолжительным,
поскольку трансфецированные клетки
постепенно погибали, либо
переставали функционировать. Не
нужно забывать, что на их
поверхности остаются фрагменты аденовируса, которые
являются мощными антигенами.
Существуют также проблемы с
устойчивой длительной экспрессией
трансфецированных генов.
У больных СГ
отчетливо прослежена взаимосвязь
между высоким уровнем ЛНП в плазме
крови и формированием атероскле-ротических бляшек в
коронарных артериях (Рис 2). Из-за
сниженного клиренса в печени все
пищевые липопротеиды очень низкой
плотности (ЛОНП) успевают
превратиться в ЛНП, которые
остаются циркулировать в
кровотоке. У здоровых людей лишь
половина ЛОНП превращается в ЛНП. У
больных концентрация и время
пребывания ЛНП в плазме крови
нарастают. В результате возрастает
число частиц ЛНП, которые
фильтруются в интиму артерий. В интиме часть ЛНП
подвергается окислению и
преобразованиям в
модифицированные частицы (м-ЛНП). Некоторые мЛНП вызывают
образование новых антигенов и
стимулируют их представление
макрофагами. Немаловажно и то, что
мЛНП приносят в интиму чужеродные
антигены (липополисахаридызапускает
бактерий, ксенобиотики). Активация макрофагов
воспалительную и иммунную
активацию эндотелия и приводит к
повышению экспрессии молекул
адгезии (ICAM, VCAM, E-selectins) на
поверхности эндотелиальныхадгезинов и клеток,
контактирующих с кровью. Активация
медиаторов хемотаксиса привлекает
новые популяции моноцитов и
лимфоцитов. Активированный
эндотелий и макрофаги интиму секретируют
в
ростовые факторы и пролиферацию
гладкомышечных клеток (ГМК), синтез экстра-целлюлярного цитокины, ускоряющие
миграцию и матрикса (фиброз) и
отложение новых порций ЛНП и мЛНП
(липоидоз). Этот "порочный
круг" повторяется в интиме много раз с
усилением, что вызывает рост
атеросклеротической бляшки.
Нерегулируемый рост бляшки в
интиме напоминает феномены "клеточного
хаоса", которые
наблюдаются при циррозе печени,
доброкачественной гиперплазии
простаты и других клеточных дисплазиях (4). В настоящее
время единственным способом
блокирования роста бляшек является
их химическое или физическое
удаление, либо механическое раздавливание (ангиопластика), а единственным
эффективным лечением гомозигот - пересадка
печени, которая восстанавливает
нормальный круговорот пищевого
холестерина в организме. Ловастатин на 20-60% снижает
уровень ЛНП у ге-терозигот, селективно
повышая уровень ЛНП-рецепторов в печени.
Однако не менее половины пациентов
плохо переносят ловастатин (это
лекарство назначается до конца
жизни, т.к. прекращение его приема
приводит к 2-3-кратному повышению
холестерина плазмы крови).
Результаты многочисленных
проектов "генной терапии" СГ пока также не
обнадеживают. Технологию пересадок
ген-трансфецированных гепатоцитов удалось
частично отработать на кроликах Ватанаби (эти кролики
оказались весьма точной гено- и фенокопией СГ человека), но
генная терапия СГ не станет в
ближайшее время клинической
реальностью. Что удерживает
родителей и врачей от этой дорогой
и весьма рискованной операции?
Во-первых, у тяжело больного
ребенка нужно под [beep]зом иссечь
треть печени, чтобы выделить около
10 млрд гепатоцитов. На этапе
изолирования и получения первичной
культуры гепатоцитов теряются
около 3 млрд клеток. Оставшиеся 7
млрд гепатоцитов ш уиго модифицируют,
используя в качестве вектора мутантный аденовирус. В качестве
"начинки" используют два гена -
ЛНП-рецептор и ген бета-галактозидазы
(активность
которого в клетках можно
контролировать с помощью хромогенного субстрата).
Максимальная эффективность трансфеции in vitro равна 40-30%.
Следовательно, пациенту вновь
возвращаются в печень только 3.5
млрд клеток с восстановленным ЛНП-рецептором от исходных 10
млрд клеток. Однако самым слабым
звеном лечения является последний
этап возвращения гепатоцитов в
печень шприцевой инъекцией.
Эффективность приживляемости трансфецированных клеток в печени
больных остается на уровне 1-2%, т. е. вся операция
завершается возвращением в печень
пациента 35 млн "вылеченных"
клеток. Остальные гепатоциты быстро
элиминируются из кровотока и
погибают, если даже после операции
использовать иммуносу-прессанты. Таким образом,
эффективность "генной
терапии" пока выражается
следующим отношением: одна
"вылеченная" клетка на 1000
"невылеченных". По этой
причине в США, Канаде, Франции и
Англии проведены только несколько
трансплантаций гепатоцитов после
их "генной коррекции" in vitro. У
всех пациентов сразу после
операции отмечался гиполипидемический эффект, который
сохранялся несколько недель.
Однако он был непродолжительным,
поскольку трансфецированные клетки
постепенно погибали, либо
переставали функционировать. Не
нужно забывать, что на их
поверхности остаются фрагменты аденовируса, которые
являются мощными антигенами.
Существуют также проблемы с
устойчивой длительной экспрессией
трансфецированных генов.